2.4基因组重测序
2.4.1数据评估与指控
采用Illumina MiSeqTM分别得到的出发菌株W303-1A、F-2、A-2和B-2基因组序列图像数据,经过CASAVA碱基识别(base calling)分析转化为原始测序序列(raw reads)。由于测序得到的原始数据中含有带接头、低质量的序列,为了保证信息分析质量,使用Trimmomatic对原始数据进行过滤,得到clean数据。如表1所示,出发菌株W303-1A、F-2、A-2和B-2检测到的序列数分别为34 205 540、19 689 382、26 299 894和39 691 162 bp,经过滤处理后,有效数据均超过了95.0%,且数据处理后,4株菌的重测序结果Q20、Q30都在95.0%以上,其中,G+C含量处于37.9%−39.7%,结果表明,测序结果有效,可用于进一步的信息分析。
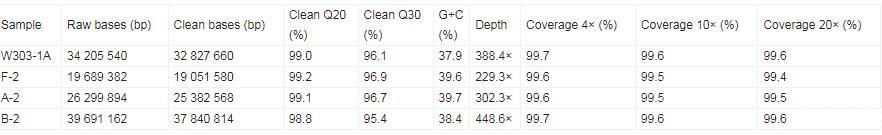
表1.重测序数据统计与评估
测序覆盖度能够间接反映变异检测的完整性,因此,平均测序深度(depth)越大,后续能够检测到的变异位点则越多。如表1所示,出发菌株W303-1A、F-2、A-2和B-2全基因组的平均覆盖深度分别为388.4×、229.3×、302.3×和448.6×,此外,4×、10×和20×的测序深度覆盖率(coverage)均为99.0%以上,这一结果进一步表明测序结果的准确性和可靠性。
2.4.2变异监测分析
如图4所示,通过主成分分析(principal component analysis,PCA)对三重复和互反实验中显示一致结果的反应微阵列数据点进行分析。出发菌株W303-1A与经驯化后菌株的差异基因分布在不同的区域,可以得出4种菌株的表达模式各不相同。其中,对羟基苯甲酸对第1个PC有正向影响,糠醛对第2个PC有负向影响。因此,A-2大致位于PC1的x轴位置。F-2位于左侧的y轴位置,A-2位于右侧象限。驯化后菌株对不同抑制环境的适应性使得部分基因的表达模式发生不同变化,这可能是因为不同驯化菌株对抑制剂的反应差异导致的。
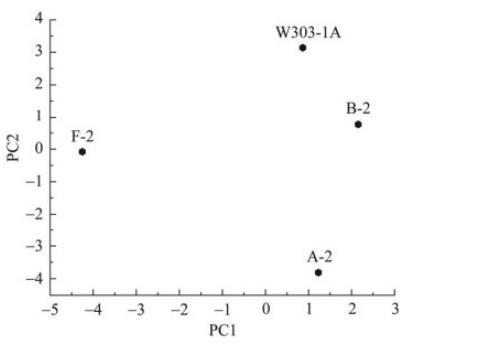
图4主成分分析
如表2所示,对4种菌株遗传变异进行分析发现,大部分突变位点分布在基因的外显子区(即CDS区域),而在UTR3以及剪切位点区域的突变数为0,这一结果表明经过驯化后,突变基因多发生在编码区,这或许是由于非编码区本身在整个基因组的比例较小。此外,与出发菌株W303-1A相比,F-2菌株具有3.7×104个SNP位点,其中在编码区的突变点有3.1×104个,同义突变点有3 397个;A-2菌株具有3.7×104个SNP位点,其中在编码区的突变点有3.1×104个,同义突变点有3 399个;B-2菌株具有3.7×104个SNP位点,其中在编码区的突变点有3.1×104个,同义突变点有4 301个。Meriem等研究表明,当基因突变位点位于编码区时,突变增加了编码蛋白序列差异的可能性,从而导致相应基因功能的改变。
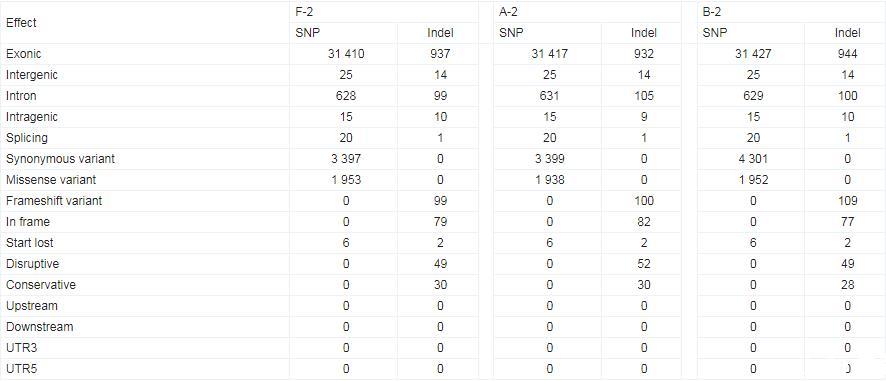
表2.SNP/Indel的注释结果
2.4.3差异表达基因的功能和代谢通路富集分析
基于GO(gene ontology)数据库对获得的SNP位点和Indel位点进行注释,确定突变基因的功能及相关描述信息,以便更加精准高效地找到目的基因。GO分析包括生物过程(biological process,BP)、细胞成分(cellular component,CC)和分子功能(molecular function,MF)。数据(false discovery rate,FDR<0.01)以条形图表示(图5A),其中,在生物过程类别下,大多数基因被注释为代谢过程和细胞过程。在细胞成分方面,差异表达基因(DEGs)主要与细胞和细胞组分有关,在分子功能类别上,大多数基因被注释为催化活性。GO功能富集分析如图5B−5D所示。其中,生物过程中含量最高是细胞缺氧反应过程(GO:0071456),其次是细胞内信号传递过程(GO:000494)和肌动球蛋白结构组织的调控过程(GO:0034497)。细胞成分中,内质网(GO:0005783)和内质网膜(GO:0005789)的含量最高。在分子功能类别中,生物素羧化酶活性(GO:0004360)是最重要的,其次是谷氨酰胺果糖-6-磷酸转氨酶活性(GO:0004075)和锌离子结合能力(GO:0008270)。
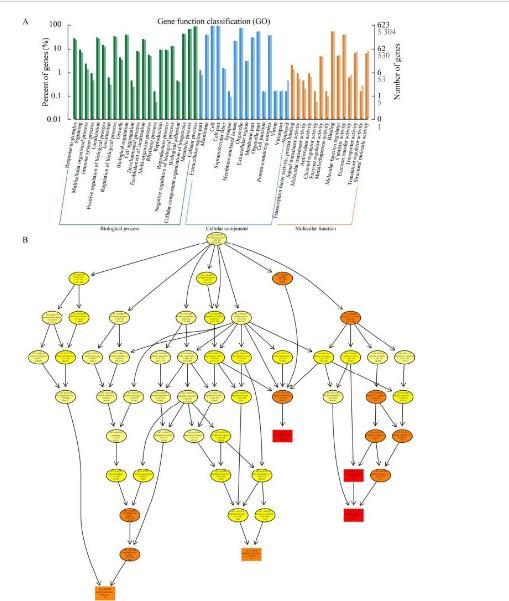
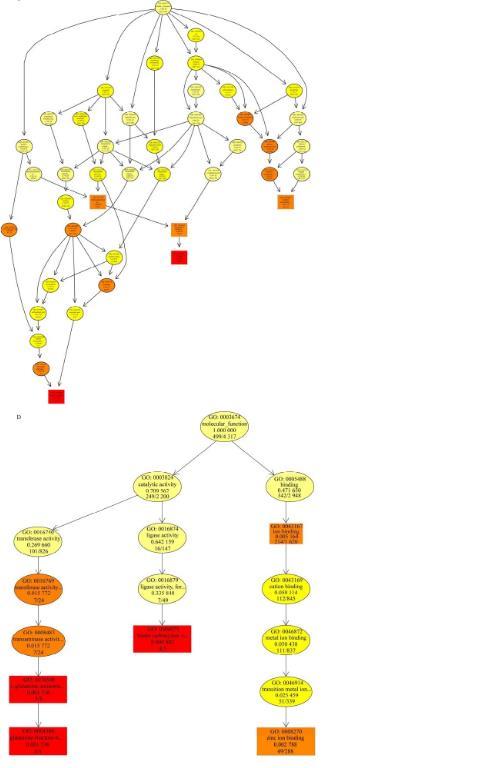
图5 GO功能注释图及富集分析。A:突变基因的GO分类;B:生物过程中突变基因的GO分类;C:细胞成分中突变基因的GO分类;D:分子功能中突变基因的GO分类析。色标为富集分析校正P值。
KEGG通路数据库是提供一个分子水平有关分子相互作用、反应和关系网络的生物系统,KEGG通路对DEGs的分类主要分为5类:代谢、生物系统、环境信息处理、细胞过程和遗传信息处理。一般来说大多数基因被注释在代谢相关的途径中,其中,代谢类别中,脂类代谢是第二类,其次是碳水化合物代谢和外源性生物降解代谢。在生物系统分类中,消化系统是最丰富的第二类。蛋白质通常不是独立行使其功能的,而是相互协调以完成一系列生化反应,因此,KEGG通路分析可以帮助揭示与疾病机制或药物作用有关的细胞过程。以KEGG数据库为参考,将158个差异基因连接到不同的路径,其中ko 00360:苯丙氨酸代谢、ko 04728:多巴胺能突触、ko 00983:药物代谢和ko 04923:调节脂肪细胞的脂质分解通路信号含量较高(图6)。通过功能富集分析可以看出,驯化后的菌株发生了一系列突变,其中参与催化活性功能的相关酶基因和参与药物代谢途径的相关基因能与菌株的耐受性密切相关,这些基因的功能注释为进一步明确驯化后菌株抗性机制提供重要素材。糠醛和对羟基苯甲酸抑制了酵母的生长或导致较长的滞后期,进而降低了纤维素乙醇的产量和生产力。根据已公布的W303-1A基因组序列,对基因组进行比对,排除匹配的遗传物质。根据SNP/Indel注释和发酵参数的统计信息,找到了与糖代谢和抑制剂抗性相关的基因(表3和表4),几种机制可以解释呋喃对乙醇发酵的抑制作用。在酵母细胞中,葡萄糖首先经过糖降解途径转换为丙酮酸,然后丙酮酸经过丙酮酸脱羧酶和乙醇脱氢酶生成乙醇。高浓糠醛进入细胞后,糠醛降解过程会与TCA循环、糖降解以及PPP等途径竞争NADH和还原酶,进而限制酵母菌生长和发酵效率。其中,在糠醛存在下生长的细胞粗提物中,与糖酵解相关的己糖激酶和甘油醛-3-磷酸脱氢酶的活性下降,使得乙醇脱氢酶中的NADH−和NADPH−进行特异性还原HMF和糠醛而被消耗。此外,糠醛可以损伤DNA,阻碍RNA和蛋白质的合成,降低酶活性,从而抑制细胞生长,同样,酿酒酵母中的多种脱氢酶/还原酶能够将糠醛和HMF还原为相应的毒性较低的醇。此外,也有研究分析表明,在酿酒酵母中,糠醛会导致活性氧积累、液泡和线粒体膜损伤、染色质和肌动蛋白损伤。通过重测序分析发现,进化菌株F-2/B-2中编码乙醇脱氢酶(alcohol dehydrogenase,ADH)、果糖-1,6-二磷酸醛缩酶(fructose-1,6-diphosphate aldolase,FBA)和丙酮酸脱氢酶(pyruvate dehydrogenase,PDH)的基因发生部分突变,这些基因与酵母的糖代谢途径相关,而脱氧酶/还原酶基因的过表达增加了糠醛还原酶的活性,从而增加了酵母对抑制剂的耐受性。Liu等的研究表明,与野生型相比,过度表达醛还原酶基因的酿酒酵母NRRL Y-12632不仅对糠醛(20 mmol/L)和HMF(40 mmol/L)的耐受性更高,而且更容易恢复,生长更佳。
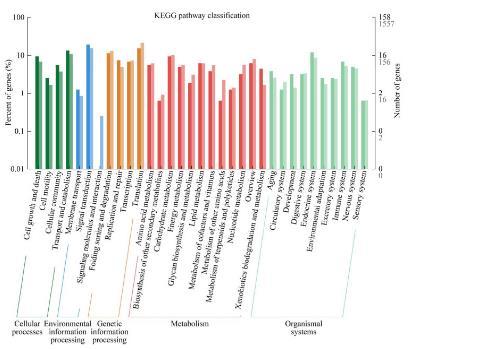
图6 KEGG通路突变基因的分类
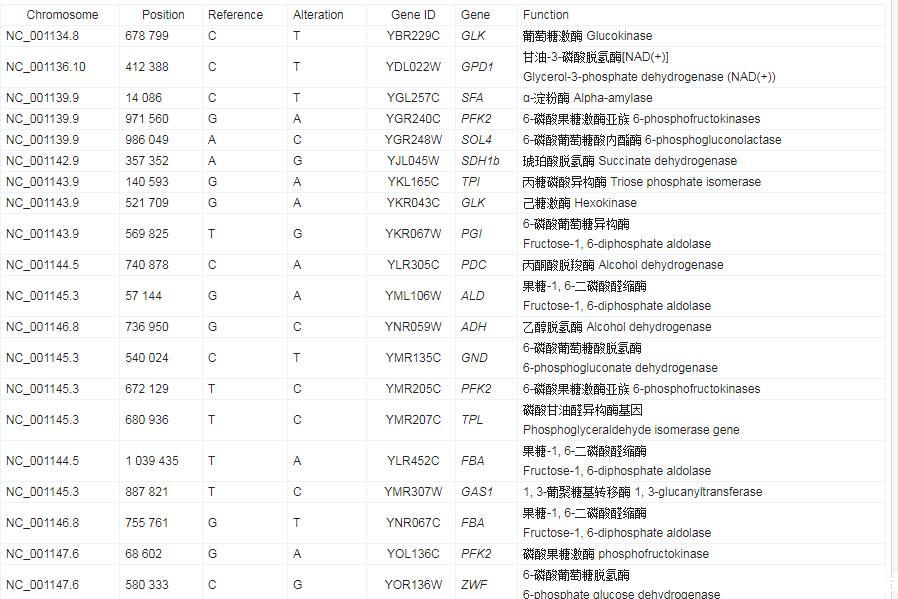
表3.与糖代谢相关主要基因的注释结果
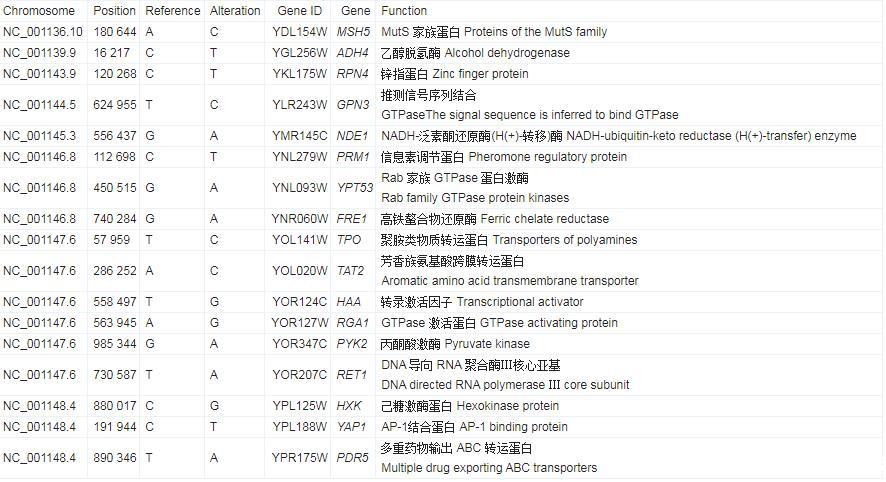
表4.与抑制物抗性相关主要基因的注释结果
酵母耐受对羟基苯甲酸的生理机制与维持细胞内pH有关,这是由质子-ATP酶泵提供的,它以ATP为代价从细胞质中去除质子。在糖酵解过程中,己糖激酶、磷酸果糖激酶和烯醇酶在弱酸和呋喃衍生物存在下的抑制已经被报道。对羟基苯甲酸在酿酒酵母中引起氧化应激,抑制蛋白质合成,损伤DNA,可能是酿酒酵母细胞生长受到抑制的原因,同时也会干扰胞内TCA循环、糖降解以及PPP等途径中酶的活性,影响菌株生长和代谢速率。通过重测序结果和相关基因的代谢通路相结合,将酿酒酵母中与抗抑制物相关的基因与已报道的相关基因进行综合分析与比较,初步确定了与抑制物相关的YAP1(参与氧化应激反应和氧化还原稳态的转录激活剂)、PDR5(耐多种化学物质的多效ABC外运载体)和RPN4(锌指蛋白)基因产生部分突变或许是导致酿酒酵母中抑制因子耐受性的主要原因。
3结论
驯化策略对于构建工业化纤维素乙醇发酵菌株十分有效。本研究通过逐渐提高压力值-抑制物浓度的策略显著提高了菌株的抑制物抗性,有效提高了乙醇生产效率。在糠醛和对羟基苯甲酸同时存在的情况下,进化菌株的最终糖转化率为98.4%。然而在实际预处理水解液中,抑制物的组成更为复杂多样;并且在纤维素乙醇生产的发酵过程中,pH值、温度以及五碳糖的利用都是影响发酵效率的重要因素。因此,提高菌株对多重胁迫的适应性才能更大限度地提升生产效率。本研究还对出发菌株和驯化后菌株进行了基因组重测序,结合参考文献耐抑制物数据对变异点进行分析挖掘,发现与抗抑制物有关的潜在突变位点,并对潜在基因进行了分析,为后续菌株构建提供更多可能的操作靶点。
作者贡献声明
樊美杉:研究构思和设计、论文撰写和修改;卢圣捷:数据收集和处理;张红丹:协助实验操作;钟春梅:提供技术支持;谢君:研究构思和设计。
相关新闻推荐
3、活性酵母与酵母培养物饲喂育肥牛生长性能、胴体指标和牛肉品质的比较