来自山东大学齐鲁医院的研究团队在《Advanced Biology》发表的最新研究揭示了肠道微生物群与克罗恩病(CD)之间的紧密联系及独特作用机制。这一发现挑战了传统对CD发病机制的认知,首次证实可从RNA-seq数据中同时恢复宿主基因表达和微生物定量信息,明确了微生物组成变化与宿主基因表达异常的潜在因果关系。研究不仅为理解肠道微生物与宿主免疫的相互作用提供了新视角,还为开发基于微生物的CD诊断方法和靶向治疗策略奠定了理论基础。
研究背景
在克罗恩病(CD)的诊疗过程中,明确发病机制并实现精准诊断始终是关键难题。肠道微生物失调与宿主基因表达异常在CD的发展中起着重要作用,然而,二者之间的因果关系一直难以明确。现有研究大多聚焦于粪便微生物,对组织驻留微生物的了解有限,且缺乏能够同时获取微生物定量和宿主基因表达数据的有效方法。传统的诊断模型大多依赖宿主基因,容易受到技术偏差和批次效应的影响,难以实现精准诊断。
转录组分析宿主-微生物相互作用(TAHMC)技术,作为一种新兴的研究手段,具备从批量RNA-seq数据中同时恢复宿主基因表达和微生物定量信息的潜力。但此前该技术在克罗恩病研究中的应用较少,其在揭示肠道微生物与宿主基因表达关系方面的能力尚未得到充分挖掘。部分研究对其数据处理和分析方法存在质疑,认为可能存在误差影响研究结果的准确性。研究团队以探究克罗恩病中组织驻留微生物与宿主基因的关联为目标,试图借助TAHMC技术解开这一谜团。
主要结果
1.TAHMC技术对肠道微生物群的精准解析与污染过滤验证
为明确克罗恩病(CD)黏膜组织微生物组成及排除数据污染干扰,研究团队利用TAHMC技术对1855例RNA-seq样本(1554例CD、301例对照)进行深度分析。通过五步过滤法(批次、相关性、丰度、黑名单及文献校验)去除污染物,最终获得692个属的黏膜共生微生物数据(47794497条reads),其中变形菌门污染物占比最高(554个属),验证了污染过滤的必要性。
对比CD与对照组发现,CD组微生物α多样性(Shannon/Simpson指数)显著降低,β多样性分析(PCoA)显示两组微生物组成存在显著差异(PERMANOVA,P<0.0001)。门水平上,CD组拟杆菌门、放线菌门丰度升高,厚壁菌门降低;属水平鉴定出108个差异菌属(91个减少,17个增加),包括已知IBD相关菌属如Akkermansia、Faecalibacterium,及潜在新关联菌属。FISH实验证实CD回肠组织中细菌主要定位于细胞内,揭示组织驻留微生物的独特分布特征。

图1涵盖TAHMC流程、污染过滤结果及FISH验证细胞内细菌存在的TAHMC技术解析肠道微生物组成及分布

图2克罗恩病与对照组肠道微生物群差异分析
2.免疫浸润亚型鉴定及其与微生物群的关联性分析
为揭示CD免疫微环境异质性及微生物群对免疫状态的影响,筛选与临床表型相关的关键免疫亚型,研究基于MCPcounter算法评估免疫细胞浸润,通过共识聚类划分免疫亚型,并结合抗TNF耐药基因集及微生物差异分析(MaAsLin2、LEfSe)。研究将CD样本分为两种免疫浸润亚型(C1/C2)。C2亚型整体免疫细胞浸润水平显著高于C1(P<0.001),且富集抗原呈递、促炎信号相关基因(如CXCL1、CCL2),提示过度免疫激活状态。进一步结合抗TNF耐药基因集分析,发现C2亚型耐药基因表达显著升高(P<0.01),且患者诊断年龄更早(P<0.05),提示更差的临床表型。
微生物群与免疫亚型关联分析显示,C2亚型中Edwardsiella、Nocardia等3个菌属富集,而C1亚型富集38个菌属(如Dorea、Alistipes),其中16个属在对照组中高丰度。α/β多样性分析表明,C1/C2亚型微生物群落结构差异显著(P<0.05),且Lachnospiraceae家族丰度与免疫激活基因呈负相关(Spearman R<−0.3,P<0.05),揭示特定微生物通过调控免疫通路影响CD进展。
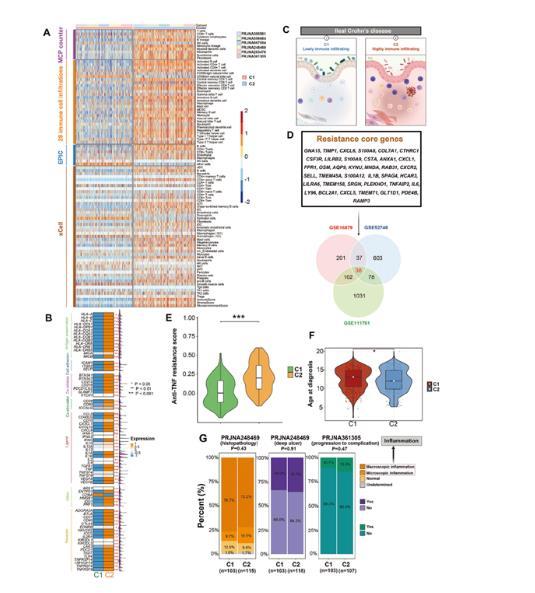
图3克罗恩病免疫浸润亚型鉴定及临床关联,呈现免疫亚型分类、免疫细胞浸润差异及抗TNF耐药性/诊断年龄关联
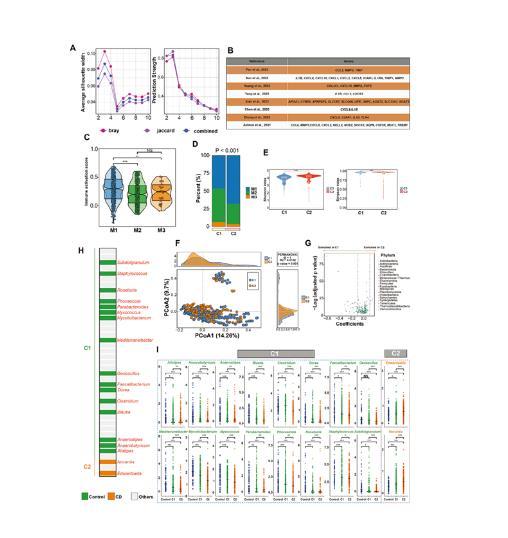
图4肠道微生物群与宿主免疫浸润关联分析,揭示微生物聚类与免疫亚型的相关性、差异菌属分布及功能通路联系
3.基因-微生物关联网络构建及诊断
模型开发为挖掘微生物与宿主基因的互作机制,构建基于微生物-基因网络的CD诊断模型,研究利用lasso回归结合稳定性选择,分别构建对照组与CD组的mRNA/lncRNA-微生物关联网络。发现CD组微生物与宿主基因的关联强度显著高于对照组(正相关边:CD组1075条vs对照组615条)。筛选差异表达基因(DEGs)与差异微生物(DAMs)的核心互作节点,构建包含11个微生物属(如Faecalibacterium、Akkermansia)和12个宿主基因(如CXCL1、BIRC3)的Host-Microbe Network Signatures(HMNSig)。
机器学习模型验证显示,HMNSig在独立测试集(ICDR队列,n=483)中诊断效能优异,AUC达0.77(95%CI:0.73–0.82),显著优于单一宿主基因或微生物模型(P<0.05)。进一步分析发现,模型核心基因富集于免疫调节、细胞黏附通路,而核心菌属多与肠道屏障功能相关,提示微生物-基因互作通过多通路影响CD发病。
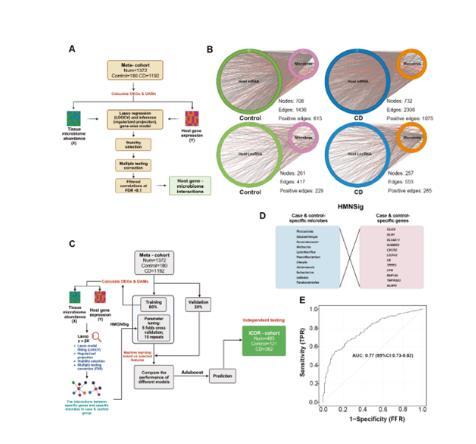
图5基因-微生物关联网络构建及诊断效能验证
4.体外实验验证微生物对宿主基因的调控作用
针对实验3中关联网络中的关键菌属Faecalibacterium prausnitzii(CD组丰度显著降低),通过Caco-2细胞共培养实验验证其对宿主基因的调控效应。转录组分析显示,共培养后44个基因显著上调(如CXCL1、CCL20,P<0.01),主要涉及趋化因子信号与炎症反应。Spearman相关性分析表明,F.prausnitzii丰度与上述基因表达呈正相关(R=0.28–0.32,P<1×10⁻⁴),与体外实验结果一致。
活/死细胞染色及细胞膜完整性检测显示,共培养后细胞凋亡率无显著变化(P*>0.05),提示F.prausnitzii通过非细胞毒性机制调控基因表达,可能与代谢产物介导的信号通路激活相关。该结果为“微生物通过宿主转录调控参与CD发病”提供了直接实验证据,支持TAHMC预测的微生物-基因互作网络的可靠性。
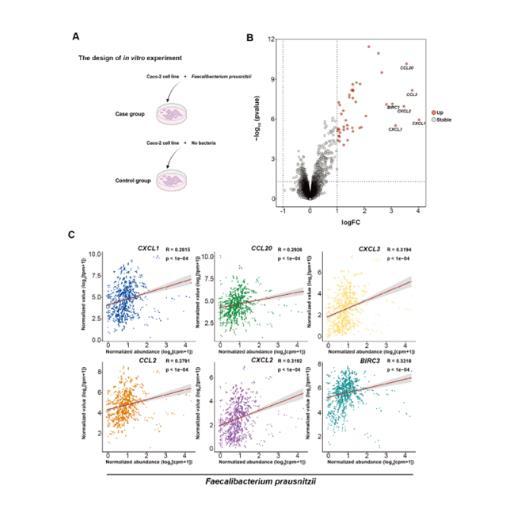
图6 Faecalibacterium prausnitzii调控宿主基因的体外验证
结论本研究通过TAHMC技术开发、微生物群深度解析、免疫浸润分型及机器学习建模等多维度研究设计,系统揭示了克罗恩病(CD)中肠道微生物群与宿主基因表达的互作机制及其临床价值。研究以CD回肠组织RNA-seq数据为核心,结合荧光原位杂交(FISH)、单细胞测序及体外共培养实验,证实TAHMC技术可从复杂数据中精准恢复微生物定量与宿主转录信息,发现CD黏膜组织微生物α多样性显著降低、关键菌属(如厚壁菌门减少、拟杆菌门增加)与免疫浸润亚型(C1/C2)密切相关,且微生物-基因关联网络在CD组中呈现更强的互作信号。研究首次阐明,CD中组织驻留微生物通过调控宿主免疫通路(如趋化因子信号、炎症反应基因)参与疾病进展,且这种互作可通过机器学习转化为高效诊断模型——包含11个微生物属与12个宿主基因的HMNSig模型在独立队列中诊断AUC达0.77,突破了传统依赖单一宿主基因的诊断局限。
体外实验进一步验证,关键菌属Faecalibacterium prausnitzii通过非细胞毒性机制上调宿主免疫调节基因(如CXCL1、CCL20),揭示微生物-基因互作的直接功能关联。该研究不仅证实了TAHMC技术在解析微生物-宿主互作中的独特优势,更首次建立了从黏膜微生物组成到宿主基因表达异常的因果链条,为理解CD发病机制提供了“微生物-免疫-基因”三轴联动的新视角。其发现的天然抑菌机制(对应丝素蛋白研究中的天然属性)转化为临床应用潜力——基于微生物-基因网络的精准诊断模型、靶向关键菌属的免疫调节疗法,为开发无化学干预、聚焦宿主-微生物稳态的新型CD治疗策略开辟了创新路径,尤其在精准医疗时代展现出通过调节微生物-基因互作实现个体化诊疗的广阔前景。
相关新闻推荐
2、不同硝酸盐浓度下渤海和黄海聚球藻生长曲线、色素含量变化——摘要