针对医疗器械抽检因“内毒素限量”和“微生物限度”不合格的问题,企业需从全流程管控入手,结合两类指标的特性(内毒素源于革兰氏阴性菌细胞壁成分,微生物限度关注活菌数量),重点从质量体系各环节强化微生物控制。
1、医疗器械企业面临各种抽检中的痛点:细菌内毒素限量及微生物限度
2023年国家药监局通报,某企业生产的血液透析浓缩物因“内毒素限量”“微生物限度”不合格,可能引发患者败血症或内毒素血症;
2022年某省抽检一次性注射器,因“细菌内毒素”超标被召回。
因不同的医疗器械风险级别不同、各国的监管限值不同,各种操作操作方法也不同,以下仅作示例:
微生物限度:非无菌产品(如贴敷类器械)需检测菌落总数、大肠菌群等指标,参考《中国药典》1105标准。
·内毒素检测:使用鲎试剂法,血液接触类器械需控制内毒素≤0.25EU/mL2。
(一)细菌内毒素的定义
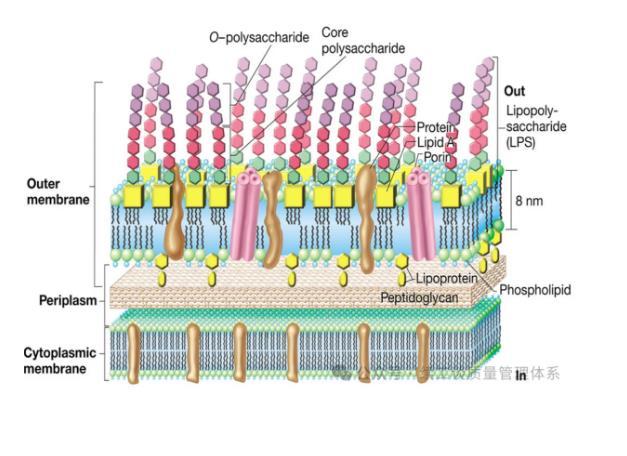
细菌内毒素是革兰氏阴性菌(如大肠杆菌、沙门氏菌等)细胞壁的主要成分——脂多糖(Lipopolysaccharide,LPS),由脂质A、核心多糖和O-特异性多糖组成。当细菌死亡或裂解时,细胞壁破裂释放内毒素,其化学性质稳定,耐高温(需160℃以上干热2小时或湿热灭菌更高条件才能破坏),且无法通过常规灭菌(如湿热灭菌、辐照灭菌)完全去除,只能通过热破坏、吸附(如活性炭)、超滤等方法降低其含量。
(二)细菌内毒素对人体的危害
内毒素进入人体后,会激活免疫系统产生过度炎症反应,其危害程度与进入体内的剂量和医疗器械接触人体的风险等级(如是否直接接触血液、体液或植入体内)密切相关。主要危害包括:
(1)发热反应
1.内毒素通过刺激巨噬细胞释放白细胞介素-1(IL-1)、肿瘤坏死因子(TNF-α)等细胞因子,导致体温调节中枢异常,引发发热(常见于输液、透析等接触体液的器械)。
(2)内毒素血症与脓毒症
高剂量内毒素进入血液后,可引发全身性炎症反应综合征(SIRS),表现为寒战、低血压、器官功能障碍,严重时发展为脓毒症休克,甚至死亡(尤其威胁免疫力低下患者或植入类器械使用者)。
(3)局部组织损伤
若器械接触局部组织(如手术缝线、导管),内毒素可诱发局部炎症、脓肿或组织坏死,延长愈合时间,增加感染风险。
(4)干扰医疗操作效果
1.对于生物检测类器械(如体外诊断试剂),内毒素污染可能干扰检测结果的准确性;对于植入材料(如人工血管、关节假体),内毒素残留可能影响材料相容性,导致植入失败。
(5)耐热性带来的控制挑战
内毒素具有极强的耐热性(需250℃干热30分钟才能完全破坏),常规湿热灭菌(121℃/15分钟)仅能杀灭细菌,但无法彻底去除已存在的内毒素,因此需通过原料管控、生产过程清洁验证和终端内毒素检测(如鲎试验)确保安全。
(三)医疗器械相关法规对细菌内毒素的限制
国家药监局及国际标准对医疗器械内毒素限量有明确要求,例如:
中国药典(四部):规定注射用器械、接触血液/体液的器械需进行内毒素检测,限值通常为≤0.5EU/mL(具体根据器械类型和临床使用剂量计算,如公式:内毒素限值=K×M/V,K为人体最大耐受量,M为器械使用剂量)。
ISO11737-1:《医疗器械微生物控制》标准中,明确了内毒素检测方法(凝胶法、动态浊度法、显色基质法)和可接受标准。
GB/T14233.2:《医用输液、输血、注射器具检验方法第2部分:生物试验方法》规定了输液器等器械的内毒素限量及检测流程。
2、内毒素和微生物在医疗器械领域的污染途径
1.原料或辅料:如天然生物材料(胶原、明胶)、未严格管控的高分子聚合物或生产用水(如纯化水系统污染)。
2.生产环境:洁净区微生物(尤其是革兰氏阴性菌)滋生,或设备、器具清洁不彻底。
3.包装与储存:包装材料密封性不足,储存过程中受污染(如潮湿环境促进细菌繁殖并释放内毒素)。
相关新闻推荐
1、纤维二糖利用基因簇对猪源巴氏链球菌生长曲线及毒力的影响——摘要、引言
2、生长曲线分析仪研究噬菌体φR1-37对宿主细菌小肠结肠炎耶尔森菌的感染动力学
3、缺氧诱导蛋白FadA:连接分枝杆菌代谢与宿主表观遗传调控的新桥梁