质粒构建、缺失和酵母转化
| 引物 | 序列(5'→3') | 限制性酶切位点 |
|---|---|---|
| 用于质粒构建 | ||
| CA1-U | CGGAATTCCTCCCCTCACTCTTCCCC | EcoRI |
| CA1-D | GGGGTACCGCACCCGTAGTTCATCCG | KpnI |
| CB1-U | CGCGGATCC AAGTGACACATACCAGCC | BamHI |
| CB1-D | CATGCATGCATTTACACGACAACGACC | SphI |
| Kan-U | CGGGGTACCCAGCTGAAGCTTCGTACGCT | KpnI |
| Kan-D | CGCGGATCCGCATAGGCCA CTAGTGGATC | BamHI |
| CA2-U | CGGAATTCCCCTGACTCATCAATGTTTATC | EcoRI |
| CA2-D | GGGGTACCGATCTTGATAGAAAGTGGGCC | KpnI |
| CB2-U | CGCGGATCCCATCATCCCTTTTATCAAAATAAGC | BamHI |
| CB2-D | CATGCATGCCGGTGTGGACAACAATGATC | SphI |
| 用于PCR验证 | ||
| C-S | GAAATAATAAGCCCAATCCCAC | - |
| K-S | CCCATATAAATCAGCATCCATG | - |
| K-X | GCTGGTCGCTATACTGCTGTC | - |
| C-X | CACTGGAGATACTTGGGACGC | - |
| Zeocin-U | CCCACACACCATAGCTTCA | - |
| Zeocin-D | AGCTTGCAAATTAAAGCCTT | - |
| 用于实时定量PCR | ||
| CAR1-F | GCTGTCCCGTGTCATTCC | - |
| CAR1-R | GACCTTCACCGTTTGTTTCTG | - |
| ACT1-F | CGTCTGGATTGGTGGTTCTA | - |
| ACT1-R | GTGGTGAACGATAGATGGAC | - |
本研究中使用的PCR引物均根据酿酒酵母S288c基因组序列(NCBI,http://www.ncbi.nlm.nih.gov/)设计,并列于表2。pUC19用作骨架构建重组质粒pUC-CA1B1K和pUC-CA2B2K。由于葡萄酒酵母细胞是二倍体,构建了两种重组质粒以删除CAR1基因的两个拷贝。第一个质粒pUC-CA1B1K的构建如下:用CA1-up和CA1-down引物扩增的CAR1基因上游同源片段(命名为CA1)的EcoRI和KpnI位点,克隆到pUC19克隆载体中,得到pUC-CA1质粒。用CB1-up和CB1-down引物以WY1基因组DNA为模板,通过PCR扩增CAR1的BamHI和SphI下游同源片段(命名为CB1),插入pUC-CA1的BamHI和SphI位点,得到名为pUC-CA1B1的质粒。然后,利用Kan-up和Kan-down引物从pUG6载体扩增用作G418抗性标记的KanMX标记基因序列。将KanMX标记基因序列亚克隆到pUC-CA1B1的KpnI/BamHI位点,获得名为pUC-CA1B1K的质粒(图1a)。缺失质粒pUC-CA2B2K的构建方法类似,但PCR产生的CA2和CB2片段分别与CAR1基因的上游和下游区域同源,比CA1和CB1片段更靠近CAR1基因,且分别与CA1和CB1片段不重叠(图1b)。
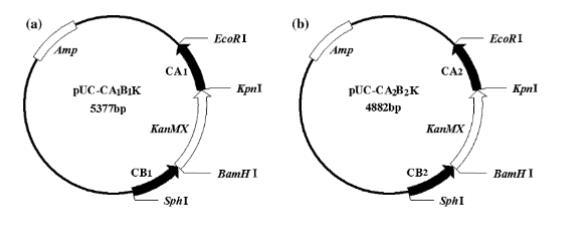
由于WY1菌株是二倍体,采用回溯引物破坏策略高效重复删除CAR1基因,如前所述。从重组质粒pUC-CA1B1K和pUC-CA2B2K分别通过PCR扩增CA1-loxP-KanMX-loxP-CB1和CA2-loxP-KanMX-loxP-CB2缺失盒。使用CA1-loxP-KanMX-loxP-CB1缺失盒删除第一个CAR1拷贝。使用Cre/loxP系统去除选择标记KanMX盒后,使用CA2-loxP-KanMX-loxP-CB2缺失盒删除第二个CAR1拷贝。然后,再次使用Cre/loxP系统去除KanMX盒。
酵母转化采用先前描述的醋酸锂方法进行。转化子在含300mg/L G418的YEPD平板上筛选。应用PCR验证具有准确位点整合的重组菌株。表2中设计了几对不同引物对。
发酵实验
将酵母细胞在5mL YEPD培养基中30°C、180rpm培养12小时,然后将细胞接种到50mL YEPD培养基中,30°C、180rpm培养12小时。通过离心(5000rpm,4°C,5分钟)收集细胞,并用50mL无菌水洗涤一次。将细胞沉淀重悬于5mL无菌水中,测量OD600。用细胞悬液接种(最终OD600为0.1)无菌250mL锥形瓶,瓶中装有190mL未过滤葡萄汁。使用前将锥形瓶在121°C高压灭菌15分钟。葡萄汁(20.45°Brix,pH 3.51,SO2 80mg/L)来自中国天津汉沽。混合物在25°C下发酵,每12小时称重一次以监测发酵进程,直到每日重量损失小于0.3g,即意味着发酵结束。所有发酵均进行三次重复。
实时定量PCR和酶活性测定
使用酵母RNA试剂盒(美国威斯康星州麦迪逊市Omega公司)提取酵母mRNA。使用带有ROX(实时PCR参比染料;中国CWBIO公司)的Ultra SYBR两步RT-qPCR试剂盒,通过实时定量PCR(RT-qPCR)评估CAR1基因表达。表达水平相对稳定的肌动蛋白基因(ACT1)用作参比基因。用于扩增目标基因CAR1和参比基因ACT1小片段的引物列于表2。使用ΔΔCt方法分析与参比菌株相比缺失菌株中CAR1转录的相对定量。
对于精氨酸酶测定,将酵母菌株在YEPD培养基中30°C、180rpm培养12小时并收集。使用Beatus Schehl方法测量精氨酸酶的活性,如所述。对于每个菌株,至少测定三个独立生物学重复和技术重复。
生长曲线测定和发酵特性分析
在YEPD培养基中培养12小时后,将酵母细胞转移到新鲜YEPD培养基中,30°C培养18小时。使用Bioscreen自动生长曲线分析仪(芬兰赫尔辛基OY Growth Curves Ab有限公司)每小时测定光密度(OD600)。使用分析天平和pH计分别测定CO2重量损失和pH值的发酵性能。发酵结束后,根据国际葡萄与葡萄酒组织的方法分析残余还原糖、乙醇和总酸等代谢物。使用高效液相色谱法结合荧光检测测定尿素。在EC分析前,将葡萄酒样品在密闭容器中67°C煮沸15分钟,如同工业操作。通过固相萃取(SPE)和气相色谱-质谱联用(GC-MS)按照国际葡萄与葡萄酒组织的描述定量EC含量。使用气相色谱(GC)对包括高级醇、酯类和挥发性酸在内的挥发性风味化合物进行定量测定。更详细的技术参数描述已发表。采用Gómez-Alonso等人报道的高效液相色谱法测定游离氨基酸。
统计分析
通过标准方差分析(ANOVA)确认重组菌株与其亲本菌株相比的统计显著性。本研究使用双尾检验。P值<0.05被认为具有统计学显著性。
相关新闻推荐
1、生长曲线分析仪研究食窦魏斯氏菌生物学共性及抑菌谱特性(三)
3、不同酵母的生长曲线、发酵力、耐冻性能测定及发酵面团感官评定(二)