NicA2和Pnao的表征
为了验证该新型基因的预测,从S16菌株克隆了nicA2并在大肠杆菌BL21(DE3)细胞中表达(文本S1)。IPTG诱导后,在大肠杆菌裂解物中发现了大约50 kDa的大量蛋白质,而在未诱导细胞或仅含有载体的细胞中观察不到这样的条带(图6A)。含有pET28a-nicA2质粒的静息细胞用于转化尼古丁,并且它们可以降解尼古丁,如UV扫描分析所示;因此证实nicA2基因产物具有预期的功能(图6B)。通过TLC和GC-MS分析鉴定了中间体N-甲基米斯明(P),并且我们观察到与先前报道相同的结果(图6C和图6D)[11]。
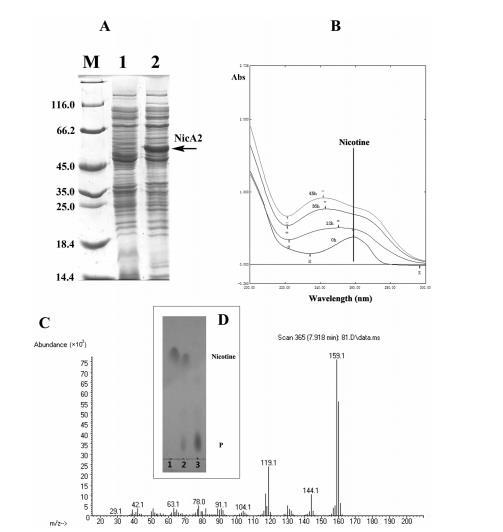
用Ni-NTA亲和柱纯化了His6标记的NicA2,并通过SDS-PAGE确认了酶的纯度,在接近50 kDa处观察到单一条带(图S5A)。进一步的实验证据支持FAD参与NicA2的功能。在382和452 nm处的紫外可见最大吸收峰,在410 nm处有最小吸收峰,是黄素蛋白的特征(图S5B)。分别敲除了nicA2和pnao基因,并进行了相关的细胞生长和静息细胞反应(文本S1)。nicA2和pnao基因缺失突变体不能在尼古丁培养基平板和液体培养物中生长(图7A和图7C)。
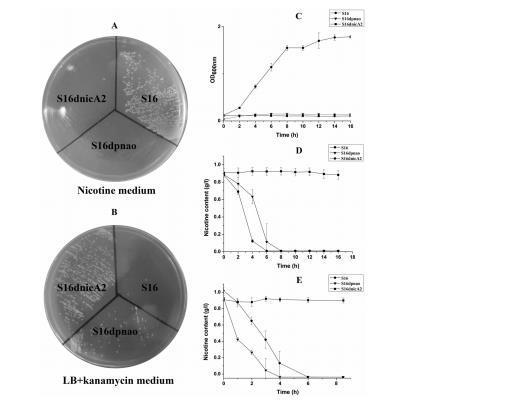
然而,缺失突变体S16dpnao在含有1 g l-1尼古丁的LB培养基中可以很好地降解尼古丁,其速率与野生型菌株S16相当(图7D)。与S16菌株相反,nicA2基因缺失突变体的静息细胞不能降解尼古丁(图7E)。pnao基因缺失突变体可以在含有1 gl-1尼古丁的LB培养基中生长并将其颜色变为深黄色,而nicA2缺失突变体的培养基颜色没有变化。此外,pnao基因缺失突变体的静息细胞降解尼古丁,但它不能进一步降解N-甲基米斯明,并且不能利用尼古丁作为唯一碳源和氮源(图7A、7C和7E)。上述事实表明酶NicA2和Pnao对于该菌株的尼古丁降解至关重要。
材料与方法
化学品
L-(-)-尼古丁(纯度99%)购自Fluka Chemie GmbH(Buchs Corp., Switzerland)。测序级胰蛋白酶来自Promega(Madison, Sweden)。SP(98%)来自Toronto(Canada)。所有其他试剂均为分析级并可商业获得。
细菌菌株、培养条件和测定
恶臭假单胞菌S16在含有1 g l-1尼古丁作为碳源和氮源的培养基中培养,如前所述[9]。该细菌也在含有1 g l-1甘油、1 g l-1 (NH4)2SO4和矿物盐培养基中培养,初始pH为7.0,含有13.3 g l-1 K2HPO4·3H2O、4 g l-1 KH2PO4、0.2 g l-1 MgSO4·7H2O和0.5 mg微量元素溶液。微量元素溶液(每升0.1 M HCl)包含以下材料和数量:0.05 g CaCl2·2H2O、0.05 g CuCl2·2H2O、0.008 g MnSO4·H2O、0.004 g FeSO4·7H2O、0.1 g ZnSO4、0.1 g Na2MoO4·2H2O和0.05 g Na2WO4·2H2O。尼古丁、SP和HSP的定量数据通过高效液相色谱(HPLC)分析获得,根据先前的报告[11,12]。
用于HPLC-MS/MS分析的蛋白质组制备
将在尼古丁或甘油作为碳源生长的恶臭假单胞菌S16细胞分别悬浮在PBS缓冲液中。用PBS缓冲液洗涤后,将细胞重悬在缓冲液(8 M尿素,0.05% SDS,10 mM DTT,10 mM Tris,pH 8.0)中,并用液氮研磨裂解。在12,000 g(10分钟,4°C)离心后,将上清液与丙酮(预冷)按体积比1:4混合。在-20°C过夜孵育后,将混合物在12,000 g(10分钟,4°C)离心。用预冷丙酮洗涤沉淀三次,并重悬在含有6 M Gu-HCl,100 mM Tris,pH 8.3的缓冲液中。使用改良的Bradford方案测定蛋白质含量。将酶溶液(100μg)悬浮在含有10 mM DTT的缓冲液中,在56°C下放置0.5小时,然后在25°C下加入50 mM IAA放置40分钟。经过3 K超滤膜超滤并用100 mM NH4HCO3冲洗膜后,将溶液的pH调节至8.0-8.5。将40μg测序级修饰胰蛋白酶加入提取物中,并在37°C下温和旋转(蛋白质:胰蛋白酶比例=50:1)进行消化过夜。
2D-LC/MS和蛋白质鉴定
为了从细胞混合物中鉴定蛋白质,使用了蛋白质组学中的多维液相色谱,使用1100 LC系统。第一维开始是用真实的连续线性盐梯度(0-130分钟,2%-35%;130-135分钟,35%-90%;135-140分钟,90%;140-141分钟,90%-2%;141-180分钟,2%)从硅胶强阳离子交换柱(0.075 mm x 5 cm)和C18柱(0.075 mm x 10 cm)(Column Technology Inc.)洗脱肽段。色谱条件:缓冲液A:H2O;缓冲液B:乙腈。纳喷雾柱直接连接到LTQ Classic离子阱质谱仪(Thermo Fisher)的孔口。纳喷雾电离通过3.5 kV的喷雾电压和200°C的加热毛细管温度实现。m/z范围从400到1800。
蛋白质组生物信息学
使用蛋白质组学发现软件1.2(ThermoFisher, CA, USA)对MS或MS/MS谱进行数据库搜索。在整个色谱运行过程中收集质谱图。通过Bioworks分析质谱图。自动识别和组织高得分肽段匹配。使用来自注释的恶臭假单胞菌S16基因组的蛋白质数据库,该数据库包含总蛋白质条目。假定电荷状态Z=1且XCorr得分>2.2,或电荷状态Z=3且XCorr得分>3.75的肽段匹配被自动接受为有效。
RNA提取和实时定量逆转录PCR(RT-qPCR)
从基本培养基平板上随机挑取恶臭假单胞菌S16的单个菌落,并将新鲜过夜培养物的1:100稀释液接种到三个250 ml锥形瓶中的50 ml基本培养基(对照)和添加了1 g l-1尼古丁的基本培养基(诱导)中。分批培养在30°C下以摇动(200 rpm)培养至中期指数期(OD600 1.4)。通过14,000 g离心2分钟收获中期指数期细胞,并将沉淀在-70°C储存过夜(16小时)。使用RNAprep pure细胞/细菌试剂盒(Tiangen)从约1x10^9个恶臭假单胞菌S16细胞中提取总RNA,并通过NanoVue(GE Healthcare)定量。使用随机六聚体引物和SuperScript III逆转录酶(Invitrogen)将0.8 mg经DNase(Fermentas)处理的总RNA逆转录为cDNA。将cDNA稀释1:10,并作为qPCR分析的模板,使用CFX96实时PCR检测系统(Bio-Rad)与SYBR Green RealMasterMix(Tiangen)和qPCR引物(表S4)。使用熔解曲线和琼脂糖凝胶分析确认PCR产物的特异性。用恶臭假单胞菌S16基因组DNA的十倍稀释液构建每个引物对的标准曲线。实验通常使用恶臭假单胞菌S16的对照和尼古丁诱导培养物进行三次重复,并将每个靶基因的阈值周期(CT)值归一化到参考基因16S rRNA基因。使用2^–ΔΔCT方法计算相对表达水平,其中ΔΔCT = (CT,靶标 - CT,16S)诱导 - (CT,靶标 - CT,16S)对照[30]。
构建spmABC基因破坏的突变体恶臭假单胞菌S16dspm
通过PCR从恶臭假单胞菌S16总DNA中扩增携带5'和3'截短的spmA片段,并克隆到pK18mob的多克隆位点中,pK18mob是一种不能在假单胞菌中复制的可移动质粒。PCR引物序列如下:spmA-Sall, CCACGTCGACCAAGTTAACTGGTTATGCGAC 和 spmA-EcoRI, CCACGAATTCAGTCCTTGGCCGAAACTTTGC。通过双亲滤膜接合[31]将重组质粒从广宿主范围动员菌株大肠杆菌S17-1转移到恶臭假单胞菌S16。供体和受体分别在37°C和30°C的LB肉汤中培养至OD600mm 0.6。将10^9个供体细胞和2x10^9个受体细胞用0.85% NaCl洗涤三次,混合并涂布在置于LB琼脂上的0.45-μm滤膜上。平板在37°C正面朝上培养5小时,然后在30°C培养12小时。然后将细胞重悬在0.5 ml 0.85% NaCl中,并以不同稀释度铺在含有100 mg l-1卡那霉素的M9平板上,并在30°C培养。通过使用引物对spmA-Sall和pK18mob-269, GCTTCCCAACCTTACCAGAG进行PCR分析测试Kan抗性转接合子。
构建含有互补spmABC基因的质粒pME6032-spm100
使用表S5中的引物从恶臭假单胞菌S16的总DNA中扩增包含spmABC基因编码区和spmA ATG上游100 bp的4.45 kb片段,然后克隆到pME6032[32]中,生成重组质粒pME6032-spm100。
构建恶臭假单胞菌S16的mfs、sapd、pnao、nicA2基因破坏突变体
通过PCR从恶臭假单胞菌S16的总DNA中扩增mfs、sapd、pnao和nicA2的DNA片段,并克隆到pK18mob的多克隆位点中。PCR引物对序列如表S6所示。通过电转化转化恶臭假单胞菌S16,条件如下:将0.5-1μg DNA加入100μl恶臭假单胞菌S16的电感受态细胞中,使用Bio-Rad Gene-Pulser Xcell(Bio-Rad Laboratories, Hercules, CA)在12 kV cm-1、200 Ω、25μF下电击。
相关新闻推荐
1、海洋来源氨基香豆素增强多粘菌素B抗伯克霍尔德菌活性(二)