1材料与方法
1.1试剂
2216E肉汤、琼脂培养基均购自青岛高科技工业园海博生物技术有限公司;刃天青购自上海源叶生物科技有限公司;细菌RNA提取试剂盒购自南京诺唯赞生物科技股份有限公司;反转录试剂盒PrimeScript™RT reagent Kit、通用荧光定量PCR试剂盒TB Green®Premix Ex Taq™(Tli RNaseH Plus)、ROX plus等RT-qPCR相关试剂均购自TaKaRa公司;板蓝根、人工牛黄、甘草、冰片、猪胆粉、玄明粉均购自云南保和堂中医药有限公司;乙醇、丙二醇、碳酸氢钠均购自上海阿拉丁生化科技股份有限公司。
1.2甘胆口服液的制备
参照江厚生等方法制备甘胆口服液(GD),称取板蓝根100 g、甘草40 g、玄明粉30 g,加1 400 mL水煎煮3次,将3次煎液合并滤过,滤液浓缩至相对密度为1.15-1.20(50℃),放冷后加入500 mL乙醇,过滤并回收乙醇,加水约250 mL,静置备用。称取猪胆粉20 g、人工牛黄34 g、冰片20 g,加入乙醇100 mL使其溶解,加入上述提取液,加水至1 000 mL,搅匀滤过后即得GD,每毫升药液约含有0.244 g原生药,即浓度约为244 mg/mL。
1.3菌株培养
副溶血弧菌CICC 21617购自中国工业微生物菌种保藏管理中心(CICC),经鉴定该菌符合副溶血弧菌的生理生化特性,多次传代后仍具有溶血性,携带PirA毒力基因,且对虾类有致病性。在副溶血弧菌CICC 21617菌液中加入终浓度为20%的甘油,保存于-80℃超低温冰箱。在无菌条件下将副溶血弧菌划线接种于2216E琼脂平板上,28℃培养24 h,挑取单菌落接种于2216E肉汤培养基,28℃、150 r/min振荡培养24 h,通过比浊法测定副溶血弧菌菌液浓度。在后续实验中,用2216E肉汤培养基将菌液调至所需浓度。
1.4最小抑菌浓度(MIC)与最小杀菌浓度(minimum bactericidal concentration,MBC)测定
采用刃天青显色法测定GD对副溶血弧菌CICC 21617的MIC,在无菌超净台中取一块无菌的96孔板,在第一列各孔中加入100μL 244 mg/mL的GD,逐列用2216E肉汤培养基进行2倍梯度稀释,使药物终浓度分别为122、61、30.5、15.2、7.6、3.8、1.9、0.96、0.48和0.24 mg/mL,另设置1列作阳性对照(加入100μL 1×105 CFU/mL的副溶血弧菌菌液)和1列阴性对照(加入100μL 2216E肉汤培养基),最后在各浓度药物组和阳性对照组的孔中添加100μL 1×105 CFU/mL的副溶血弧菌菌液,阴性对照加等量的2216E肉汤培养基。将96孔板放入28℃恒温培养箱,24 h后加入50μL蓝色刃天青溶液,12 h后观察培养液颜色变化,若培养液保持蓝色表明细菌生长受抑制,培养液变粉色说明有细菌生长,将抑制细菌生长的最低药物浓度作为MIC。吸取MIC浓度以上菌液各100μL,分别涂布于2216E琼脂平板上,28℃培养24 h,观察是否有菌落长出,将无菌生长的最低药物浓度作为MBC。
1.5生长曲线测定
将副溶血弧菌CICC 21617接入2216E肉汤培养基培养过夜,并用培养基将菌液浓度调整为1×107 CFU/mL,按照体积分数为1%的接种量接入含6 mL灭菌2216E肉汤培养基的无菌试管(规格为10 mL),即得初始菌液浓度为1×105 CFU/mL。上述试管分别加入不同浓度的GD药液,使其终浓度分别为MIC、1/2MIC和1/4MIC,同时设置空白对照组(添加等量PBS溶液),每组3个重复。将试管放入全自动微生物生长曲线分析仪,在28℃、150 r/min条件下振荡培养32 h,确保达到细菌的稳定生长期,其间每2 h测定菌液OD600值,绘制细菌生长曲线。
1.6扫描电镜(SEM)和透射电镜(TEM)观察
将副溶血弧菌CICC 21617培养12 h至对数生长期(OD600值为0.6-0.8),用培养基将菌液浓度调整至1×105 CFU/mL,加入终浓度为1/4MIC的GD,另设置对照组(添加等量PBS溶液),28℃静置6 h,取1 mL菌液,在4℃、4 000 r/min条件下离心10 min,弃去上清液,用PBS清洗菌体沉淀,加入2.5%戊二醛,放入4℃冰箱过夜。用不同浓度乙醇(30%、50%、70%、90%和100%)对样品进行连续梯度脱水,采用干燥仪干燥样品,喷金制样,通过扫描电子显微镜(HITACHI公司)和透射电子显微镜(HITACHI公司)观察菌体表面及内部形态结构。
1.7转录组实验设计及细菌RNA提取
基于转录组测序技术分析GD对副溶血弧菌CICC 21617转录水平的影响。收集超过0.1 g的菌体,依据预实验结果确定初始菌液浓度为1×108 CFU/mL,GD药物浓度为1/4MIC。实验组(GD)副溶血弧菌菌液中添加1/4MIC浓度的GD,对照组(CK)添加等量PBS,每组3个重复。将两组菌液置于28℃、150 r/min条件下处理6 h,参照翟立公等方法提取菌液总RNA,使用Agilent 2100生物分析仪测定提取的RNA质量,采用NanoDrop 2000(ThermoFisher Scientific公司)超微量分光光度计测定RNA浓度和纯度,将符合质量要求(OD260/OD280为1.8-2.0,OD260/OD230大于2.0,总质量大于2μg)的样品用于后续实验。
1.8 cDNA文库构建及测序数据质控
利用反转录试剂盒将RNA反转录为cDNA,随后对cDNA进行纯化和浓缩,再依次进行末端修复、加碱基A、加测序接头处理、PCR纯化和富集等操作,从而构建出最终的cDNA文库,然后用NovaSeq X Plus平台进行测序。对下机数据进行质控处理,去除包含adapter序列的reads、剔除测序质量值低于Q20的序列等。使用统计学方法对所测得的reads进行碱基质量、碱基错误率等分析,采用Bowtie2将测序序列与副溶血弧菌参考基因组(GenBank登录号为gca_000196095.1)进行比对,获得基因的功能信息。
1.9差异表达基因及功能分析
采用韦恩图分析GD处理组和对照组的共享及特有基因;运用主成分分析(principal component analysis,PCA)表征不同组别样品转录组的总体差异;利用R语言的heatmap软件包绘制热图,对各组样品进行聚类分析。使用DESeq2筛选出各组间差异表达基因(|log2 fold change|>1,P<0.05),并通过表达量差异散点图可视化显著上调和下调的基因数量及其分布。基于Fisher精确检验方法,利用goatools软件对差异表达基因进行GO富集分析,采用KOBAS软件对差异表达基因进行KEGG信号通路的富集分析,当经过校正的P值(Padjust)<0.05时,认为此GO功能或KEGG信号通路被显著富集。
1.10逆转录实时定量聚合酶链式反应(RT-qPCR)验证
为验证测序结果的准确性,选取显著上调和下调基因各6个,进行RT-qPCR验证试验。将这些待验证的基因序列上传至NCBI数据库,使用Primer-BLAST功能设计目的基因引物(表1),并由浙江尚亚生物技术有限公司合成。采用反转录试剂盒将RNA反转录为cDNA,以此为模板,以16S rRNA基因为内参基因,进行RT-qPCR试验。反应体系(10μL):SYBR Mix 5μL,上、下游引物(10μmol/mL)各0.4μL,cDNA模板1μL,ddH2O 3.2μL。反应程序:95℃预变性10 min;95℃变性10 s,55℃退火10 s,72℃延伸30 s,45个循环。采用熔解曲线分析产物特异性。
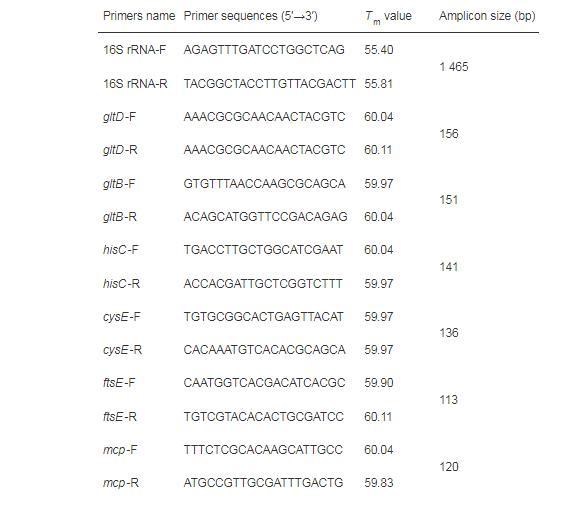
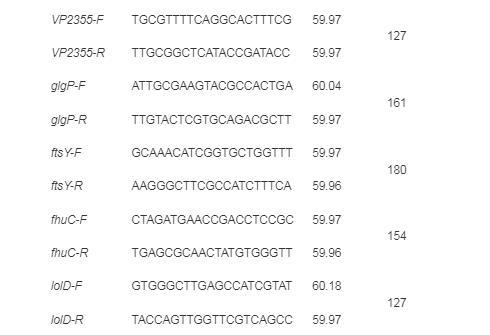
表1 RT-qPCR所用引物
1.11数据处理及分析
实验数据采用SPSS v27.0.1进行统计分析,采用单因素方差分析(one way ANOVA)或独立样本t检验(independent t-test)检验各组数据的差异,表示P<0.05,表示P<0.01。文中图表用Origin 2022或Microsoft Visio 2023绘制。
相关新闻推荐
1、Act0988放线菌拮抗菌株的鉴定、培养条件与菌株生长关系、发酵工艺(二)
3、不同浓度没食子酸对溶藻弧菌生长、电导率生、物被膜形成、泳动聚集能力的影响(一)